In artikel 26a van het Besluit ggo 2013 is de vereenvoudigde procedure voor vergunningen onder vaste voorschriften (VoV) vastgelegd voor ggo toepassingen in klinische studies waarvan op basis van ervaring en kennis van eerdere milieurisicobeoordelingen de milieurisico’s goed bekend zijn.
Op dit moment is de vereenvoudigde procedure voor een VoV van toepassing voor de klinische studies met de volgende categorieën ggo’s:
- genetisch gemodificeerde virale vectoren afgeleid van Adeno-associated dependoparvovirus A of B (AAV) zonder schadelijke sequenties (Regeling ggo Artikel 39a);
- humane cellen genetisch gemodificeerd met virale vectoren afgeleid van muizen gamma-retrovirussen dan wel humaan immunodeficiëntievirus (HIV), waarbij er geen risico is op de vorming van replicatiecompetent virus en waarbij residuele infectieuze retrovirale of lentivirale partikels afwezig zijn (Regeling ggo Artikel 39b);
- humane cellen genetisch gemodificeerd met virale vectoren afgeleid van humaan immunodeficiëntievirus 1 en gepseudoty-peerd met Vesicular stomatitis virus glycoproteïne (VSV-G), waarbij er geen risico bestaat op de vorming van replicatiecompetent virus en waarbij residuele infectieuze SIN lentivirale partikels in het medisch product aanwezig kunnen zijn (Regeling ggo Artikel 39c);
- humane cellen genetisch gemodificeerd met virale vectoren afgeleid van Adeno-associated dependoparvovirus A of B zonder schadelijke sequenties (Regeling ggo Artikel 39d).
Indien de aanvraag aan de onderstaande aanvullende voorwaarden voldoet geldt een maximale streeftermijn van 28 of 56 dagen voor de afhandeling van de aanvraag.
AAV zonder schadelijke sequenties
In artikel 40a van de Regeling ggo 2013 zijn de vereisten opgenomen voor een VoV vergunningaanvraag voor de toepassing van (Adeno-Associated Virus) in klinische studies. Van die vereisten zijn alleen f, g en h mogelijk relevant om onderscheid te maken tussen een afhandeling binnen 28 dan wel 56 dagen.
f) Gegevens betreffende de moleculaire karakterisatie van de genetisch gemodificeerde AAV vector
- 28 dagen indien de identiteit van het vectorgenoom (ITRs en tussenliggende sequenties) dan wel van de voor de vervaardiging gebruikte plasmiden in het geval van transiënte transfectie geverifieerd is middels sequencen
- 56 dagen indien de moleculaire karakterisatie op een andere manier is uitgevoerd
g) Gegevens betreffende de mogelijkheid tot vorming van replicatiecompetent AAV
- Geen noodzaak tot differentiatie
h) Gegevens over de afwezigheid van infectieus helpervirus
- 28 dagen indien geen gebruik is gemaakt van een helpervirus gedurende de productie
- 28 dagen indien gebruik is gemaakt van een wildtype helpervirus
- 56 dagen indien gebruik is gemaakt van een gg helpervirus
Ex vivo lentiviraal/retroviraal getransduceerde cellen zonder residuele infectieuze retrovirale of lentivirale partikels
In artikel 40b van de Regeling ggo 2013 zijn de vereisten opgenomen voor een VoV vergunningaanvraag voor de toepassing van ex vivo lentiviraal/retroviraal getransduceerde cellen in klinische studies. Van die vereisten zijn alleen f, h en i mogelijk relevant om onderscheid te maken tussen een afhandeling binnen 28 dan wel 56 dagen.
f) Gegevens betreffende de moleculaire karakterisatie van de genetisch gemodificeerde retrovirale dan wel lentivirale vector
- 28 dagen indien de identiteit van het vectorgenoom (LTRs en tussenliggende sequenties) dan wel van de voor de vervaardiging gebruikte plasmiden in het geval van (transiënte) transfectie geverifieerd is middels sequencen
- 56 dagen indien de moleculaire karakterisatie op een andere manier is uitgevoerd
h) Gegevens over de afwezigheid van replicatiecompetent retrovirus dan wel replicatiecompetent lentivirus
- 28 dagen voor 3de generatie SIN lentivirale systemen
- 56 dagen voor de overige lentivirale systemen en voor retrovirale systemen
i) Gegevens over de afwezigheid van residuele infectieuze retrovirale dan wel lentivirale partikels in het celproduct dat aan de proefpersoon wordt toegediend
- 28 dagen indien middels de (Commissie Genetische Modificatie) formule is berekend dat de reductieratio groter is dan 100
- 56 dagen indien de afwezigheid op een andere manier wordt berekend dan wel aangetoond
Rationale
De afgelopen jaren is met toepassing van gentherapie veel ervaring en kennis opgedaan, met name als het gaat om de veiligheid van het "platform" waarmee de feitelijke therapeutica worden gemaakt. Deze platforms bestaan uit ongevaarlijk gemaakte virussen die in staat zijn om zonder (al te veel) schade in de vorm van bijwerkingen een cel binnen te dringen en de benodigde wijziging in het DNA van de cel aan te brengen. Door deze opgebouwde kennis naar aanleiding van eerdere vergunningverleningen volgens de uitgebreide reguliere vergunningprocedure is de uitkomst van de milieurisicobeoordeling (hierna: MRB) voor dergelijke virussen tegenwoordig van tevoren bekend, namelijk verwaarloosbaar klein. Dat maakt dat er voor dergelijke ggo toepassingen geen noodzaak meer is voor een intensieve beoordeling om de veiligheid te garanderen, waardoor de beslistermijn ingekort kan worden. Met de constatering dat de uitkomst van de MRB altijd op «verwaarloosbaar risico» uitkomt, is ook de noodzaak van publieke consultatie vooraf overbodig geworden. Voor dergelijke ggo-toepassingen is de vereenvoudigde procedure in het Besluit ggo 2013 opgenomen middels artikel 3.26a.
Op basis van de genoemde ervaringen zijn voor enkele ggo-toepassingen van gentherapeutica inmiddels algemeen geformuleerde, breed toepasbare MRB’s opgesteld. Binnen de reikwijdte van de MRB (de scope) zal altijd sprake zijn van verwaarloosbaar risico. Aanvragers hoeven voor de ggo-toepassingen die binnen de reikwijdte vallen van artikel 3.26a geen individuele MRB uit te voeren, maar verwijzen naar de gestandaardiseerde MRB. Daarmee wordt het opstellen van de aanvraag eenvoudiger en is deze dus sneller gereed. Omdat de beoordeling door het bevoegd gezag eenvoudiger is, ontvangt de aanvrager ook eerder de vergunning en kan hij eerder met de klinische studie starten.
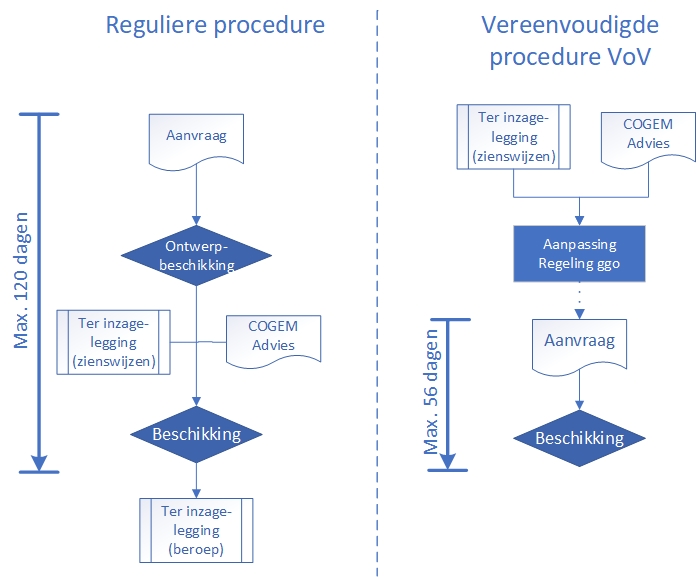
In de onderstaande figuur is het procedurele onderscheid tussen de reguliere en de vereenvoudigde procedure schematisch weergegeven.
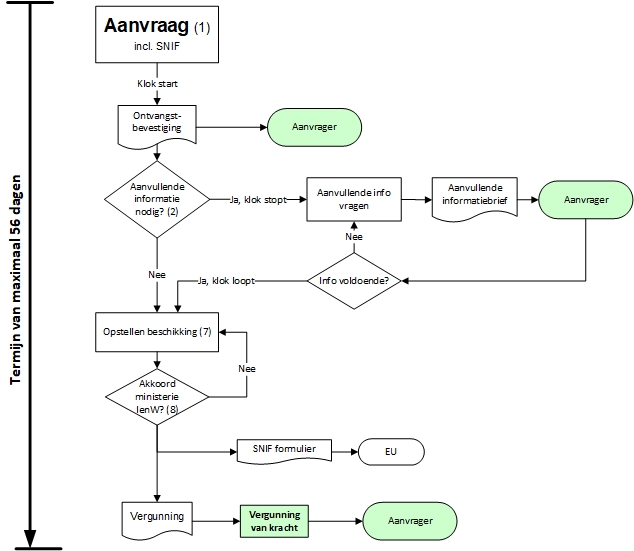
De procedurele afhandeling van de vergunningaanvraag onder vaste voorschriften
De vergunningaanvraag voor gentherapieonderzoek moet worden ingediend bij het Ministerie van IenW (p/a Bureau (Genetisch Gemodificeerd Organisme))(1) via het digitale portaal van Bureau GGO (DIGV portaal) . Volg hierbij de instructies op van het portaal en/of de beschikbare handleiding. De proceduretermijn start op het moment dat aanvraag is ontvangen. Tevens wordt een ontvangstbevestiging verstuurd. De proceduretermijn kan stopgezet worden zodra verzocht wordt aanvullende informatie beschikbaar te stellen. De termijn zal vervolgd worden zodra de gevraagde aanvullende informatie is ontvangen (2).
Behalve een vergunningaanvraag bij het ministerie van IenW moeten de voorgenomen werkzaamheden ook gemeld worden aan de andere lidstaten van de Europese Gemeenschap. Het hiervoor beschikbare SNIF (Summary Notification Information Format) formulier moet ingediend worden via het E-Submission Food Chain Platform (EFSC).
Binnen acht weken moet een beschikking opgesteld worden (7) die door het ministerie van IenW wordt ondertekend (8). De beschikking wordt naar de rechtspersoon opgestuurd. Nadat de vergunning verleend is wordt deze direct van kracht.
In de onderstaande figuur is het procedurele afhandeling van de vereenvoudigde procedure schematisch weergegeven.